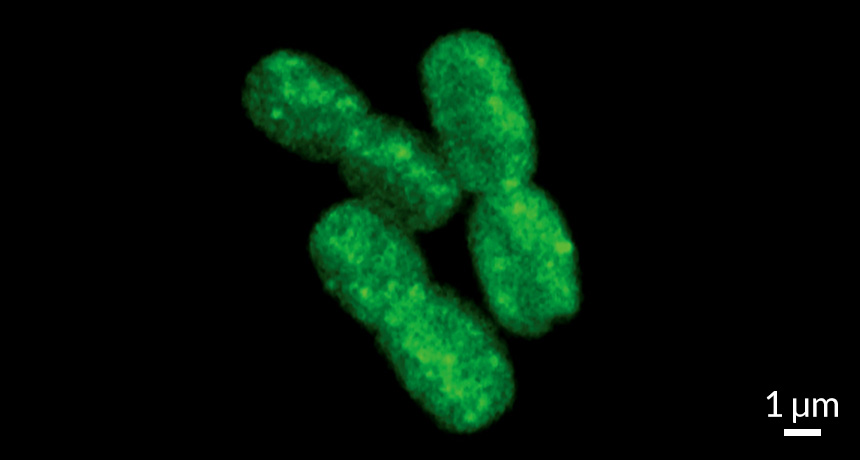
When plankton on the high seas catch a cold, the whole ocean may sneeze. Viruses hijacking these microbes could be an important overlooked factor in tracing how living things trap — or in this case, fail to trap — the climate-warming gas carbon dioxide.
Plants and other organisms that photosynthesize use energy from the sun to capture CO2 for food. The most abundant of these photosynthesizers on the planet are marine cyanobacteria with hardly any name recognition: Synechococcus and Prochlorococcus. Now, for the first time, a study looks in detail at what happens when some of the abundant viruses found in the sea infect these microbes. Two viruses tested in the lab hijacked cell metabolism, allowing photosynthesis to continue but shunting the captured energy to virus reproduction. The normal use of that energy, capturing CO2, largely shuts down, David Scanlan of the University of Warwick in England and colleagues report online June 9 in Current Biology. As a result, people could be overestimating by 10 percent the amount of CO2 that photosynthesis in the oceans captures.
On any given day, 1 to 60 percent of these plankton may have picked up a viral infection, researchers have estimated. That means that between 0.02 and 5.39 petagrams of carbon — up to 5.39 billion metric tons — may not be captured by marine organisms a year. The high end of that scenario is equivalent to 2.8 times the CO2 routinely captured annually by all the planet’s salt marshes, coral reefs, estuaries, sea grass meadows and seaweeds put together.
Synechococcus and Prochlorococcus plankton “are organisms that you’ve never heard of but you really should have,” says Adam Martiny of the University of California, Irvine. He studies the same kinds of plankton but wasn’t involved in the new virus research, and what he appreciates about it is the intriguing biology of viral manipulation the new work has uncovered.
Until now, Scanlan says, the prevailing view was that while infected plankton were still alive, they were probably carrying on normal photosynthesis. As early as 2003, researchers had clues that the viruses attacking these tiny marine organisms might manipulate photosynthesis in some way, perhaps keeping the process running in an infected cell. These viruses have genes for proteins used in photosynthesis, even though a virus doesn’t even have its own cell much less a way to photosynthesize.
What the viruses are doing, Scanlan and his colleagues have now shown, is subverting their victim’s photosynthesis. Energy capture, the part of photosynthesis directly involved with light, goes on as usual; the cells carry out the routine electron transport for catching energy. But instead of using those sizzling electrons to capture CO2 and turn it into carbohydrates for basic cell metabolism, the viruses shut down this process (called carbon fixation). The light reactions are the ones that researchers normally measure to estimate how much carbon photosynthesis captures in the oceans, but the covert viral shunting means that estimate could be too high.
Scanlan cautions that this is just the beginning of working out the numbers and possible climate effects of virus diseases for these organisms. Whatever the current effects of this takeover turn out to be outside the lab, they may intensify as the climate changes. Synechococcus and Prochlorococcus are “projected to be winners in the new, warmer oceans” and may become even more numerous, Martiny says. And what’s good for them may also increase the abundance of the viral pirates that hijack them.
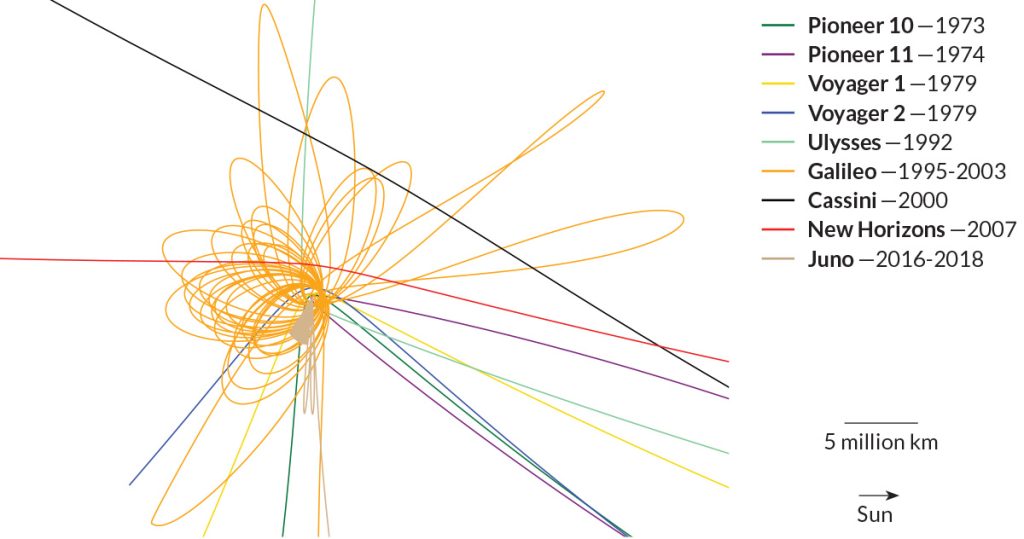
Since 1973, eight spacecraft have flown past or orbited Jupiter. On July 4, NASA’s Juno probe will become the planet’s ninth visitor.
Juno’s trajectory is different than all others, as seen in the plot above and in the video. For 20 months, Juno will repeatedly skim the cloud tops, looping over the poles on orbits that are almost perpendicular to Jupiter’s equator.
Most other spacecraft zipped by, using the planet’s gravity to speed them along to other destinations. Only Galileo, which arrived in 1995, stuck around; it spent nearly eight years circling Jupiter’s equator, repeatedly buzzing the four largest moons.
Young zebra finches (Taeniopygia guttata) learn to sing from a teacher, usually dad. Remembering dad’s tunes may even be hardwired into the birds’ brains.
Researchers at the Okinawa Institute of Science and Technology in Japan measured activity in the brains of male juvenile birds listening to recordings of singing adult males, including their fathers. The team focused its efforts on neurons in a part of the brain called the caudomedial nidopallium that’s thought to influence song learning and memory.
A subset of neurons in the caudomedial nidopallium lit up in response to songs performed by dad but not those of strangers, the team reports June 21 in Nature Communications. The more baby birds heard songs, the more their neurons responded and the clearer their own songs became. Sleep and a neurotransmitter called GABA influenced this selectivity.
The researchers suggest that this particular region of the brain stores song memories as finches learn to sing, and GABA may drive the storage of dad’s songs over others. Researchers played a variety of sounds for young zebra finches: their own song, dad’s song and songs and calls from other adult finches. Over time, their songs became more and more similar to that of their father.
Hair, scales and feathers arose from one ancestral structure, a new study finds.
Studies in fetal Nile crocodiles, bearded dragon lizards and corn snakes appear to have settled a long-standing debate on the rise of skin coverings. Special skin bumps long known to direct the development of hair in mammals and feathers in birds also turn out to signal scale growth in reptiles, implying all three structures evolved from a shared ancestor, scientists report online June 24 in Science Advances. In embryonic birds and mammals, some areas of the skin thicken into raised bumps. Since birds evolved from ancient reptiles, scientists expected that modern snakes, lizards and crocodiles would have the same structures. A study at Yale University last year found that one protein already known to be important in hair and feather development is also active in the skin of developing alligators. But the team did not find the telltale skin thickening. Without that evidence from modern reptiles, scientists weren’t sure if the bumps had been lost in reptiles, or if birds and mammals had evolved them independently, using the same set of genes. The new results are “a relief,” says Michel Milinkovitch, whose lab led the new study at the University of Geneva. Scientists had come up with a variety of complicated ideas to explain how birds and mammals could share a structure that reptiles lack. But, he says, “the reality is much simpler.”
Clues from a mutant lizard inspired Milinkovitch’s team to probe the mystery. Nicolas Di-Poï, a coauthor of the new study who is now at the University of Helsinki, found that a hair-development gene called EDA was present, but disrupted, in scaleless, or “silky,” bearded dragons. Di-Poï and Milinkovitch searched for similar molecular signals in normal reptile embryos and found genes and proteins associated with hair and feather growth studding the skin. Cell staining revealed characteristic skin thickening at those signal centers.
Reptilian skin bumps eluded previous researchers because they are tiny, appear briefly and don’t all come in at once as they do in mammals, Milinkovitch speculates. “You have to look in the right place at the right time to see them,” he says. “Then boom, you see them, and you’re like, ‘Whoa, they are exactly the same.’”
This study “addresses a fundamental question about identity for skin structures,” says paleontologist Marcelo Sánchez of the University of Zurich, who was not involved in the new research. It’s especially important that the team used crocodiles, lizards and snakes, which are far from typical lab animals, he says. Using nonmodel organisms “gives new insight into evolution we wouldn’t get otherwise.”
The next step is to understand how hairs, feathers and scales diversified from the same ancestral structure. That primordial body covering wasn’t necessarily a scale, says evolutionary biologist Günter Wagner, an author of the 2015 Yale study. “Even though intuitively you would think reptilian-like skin is ancestral, compared to mammals,” he says “it’s entirely unclear what kind of structure the scales and feathers on the one side and hair on the other has evolved from.”
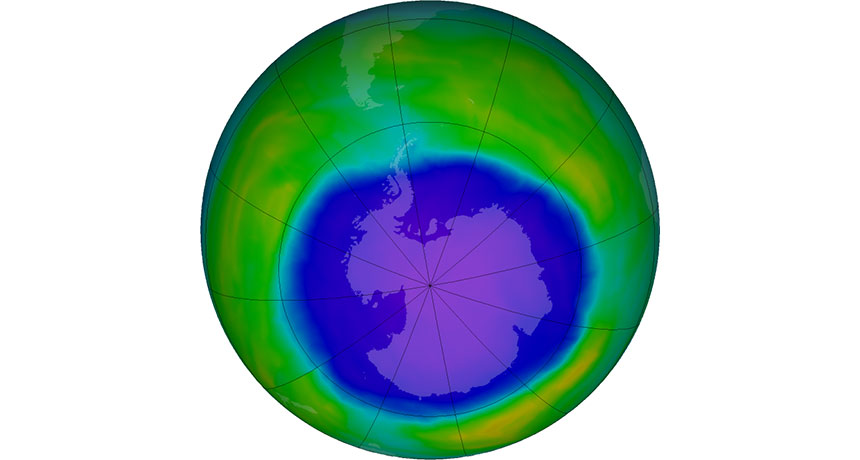
A gaping wound in Earth’s atmosphere is definitively healing. Since 2000, the average size of the Antarctic ozone hole in September has shrunk by about 4.5 million square kilometers, an area larger than India, researchers report online June 30 in Science. While the hole won’t close completely until at least midcentury, the researchers say the results are a testament to the success of the Montreal Protocol. That international treaty, implemented in 1989, banned ozone-depleting chemicals called chlorofluorocarbons worldwide.
Ozone helps shield life on Earth from hazardous ultraviolet radiation. Tracking the ozone layer’s recovery process is tricky because natural phenomena such as volcanic eruptions and weather variations can alter the size of the ozone hole. While some earlier studies suggested that the ozone had already begun healing (SN: 6/4/11, p. 15), many scientists questioned whether the work had been detailed enough to separate out the effects of natural variability.
MIT atmospheric scientist Susan Solomon and colleagues used a sophisticated 3-D atmospheric simulation to distinguish between the forces acting on atmospheric ozone. The work suggests that about half of the ozone hole’s recent shrinkage resulted from a drop in chlorofluorocarbons in the atmosphere; the remainder stemmed from weather changes.
Volcanic eruptions obscure healing signs. Last October, the ozone hole reached a record-setting average size of 25.3 million square kilometers — an area larger than Russia — thanks to the April 2015 eruption of Chile’s Calbuco volcano. That large size doesn’t disprove that the ozone hole is healing in the long run, though. Without the temporary 4.2-million-square-kilometer boost from the volcano, the hole’s average size would have peaked at a more modest 21.1 million square kilometers, the researchers estimate.
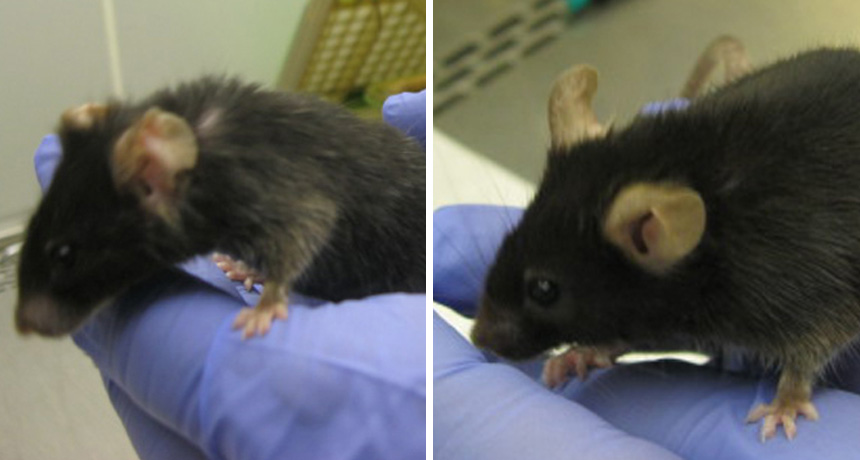
For a “three-parent baby,” getting disease-free mitochondrial DNA from a surrogate may do more than just avert disease: For better or for worse, a donor’s mitochondria could also affect the course of aging, new research shows.
Two strains of mice – genetically identical except for the source of their mitochondria, the energy centers of cells – aged very differently, researchers report online July 6 in Nature. Even though both mouse strains had healthy mitochondrial DNA, the mice with mitochondria that did not come from the same source as the rest of their DNA fared better later in life: After two years, these mice showed fewer signs of aging and had a lower incidence of tumors. The results don’t necessarily mean that a mitochondria transplant leads to a healthier life. This is just one case, researchers caution. Other DNA mixes and matches could turn out differently. But the study’s finding does point to a larger relationship between mitochondrial DNA and aging and raises new questions about the long-term effects of creating three-parent babies.
What the new results mean for people is still unclear, says Michio Hirano, a neurologist at Columbia University who was not involved in the study. But if the findings do apply to humans, he says, “you can blame your mother for how you age.”
Mitochondrial DNA is passed down from mother to child. Three-parent babies are created through an in-vitro fertilization technique that substitutes a mother’s diseased mitochondria for the healthy mitochondria of a surrogate (SN: 11/17/12, p. 5). In the procedure, which is legal in the United Kingdom and deemed ethical by a U.S. panel of experts this year (SN Online: 2/3/16), a baby inherits its nuclear DNA — the majority of its genetic fingerprint — from mom and dad. But a small amount of DNA — just 37 genes — comes from the mitochondria of a second, healthy woman.
Mitochondria do more than just power cells; they also play big roles in cell-to-cell communication and metabolism. Over the last two decades, mitochondria have also been implicated in aging but without conclusive evidence. The new research, Hirano says, “adds fuel to this debate.”
In the study, José Enríquez of the Spanish National Center for Cardiovascular Research in Madrid and colleagues bred two strains of mice. The original strain was called C57/Black 6. A second strain of C57/Black 6 carried mitochondria from another kind of mouse called NZB. This mismatch mimicked the effects of a mitochondrial transplant. Early in life, normal C57 mice bulked up faster than those carrying NZB mitochondria and had 11 percent longer telomeres (protective caps at the ends of chromosomes that get shorter over time, so are used as a proxy for aging). But later in life, the mice with NZB mitochondria had longer telomeres, less fat in their muscles and lower risk of having liver tumors at the end of their lives. Young C57 mice “tend to be stronger,” Enríquez says, probably because their mitochondrial and nuclear DNA are a good match and make efficient mitochondria. The weaker batteries in the mice with mismatched mitochondria may cause more cellular stress early on, he says, which may toughen up these mice to age more gracefully.
Since the study was done in mice, researchers don’t know how mitochondrial substitution would affect aging in humans. To avoid unforeseen and unwanted consequences, Enríquez urges caution. “Before we understand it better,” a mitochondrial transplant should mimic natural conditions, he says: “Why don’t we match the mitochondrial DNA of the donor and receptor?”
On the inevitability scale, death and taxes are at the top. Aging is close behind.
It’s unlikely that scientists will ever find a way to avoid death. And taxes are completely out of their hands. But aging, recent research suggests, is a problem that science just might be able to fix.
As biological scientists see it, aging isn’t just accumulating more candles on your birthday cake. It’s the gradual deterioration of proteins and cells over time until they no longer function and can’t replenish themselves. In humans, aging manifests itself outwardly as gray hair, wrinkles and frail, stooped bodies. Inside, the breakdown can lead to diabetes, heart disease, cancer, Alzheimer’s disease and a host of other problems. Scientists have long passionately debated why cells don’t stay vigorous forever. Research in mice, fruit flies, worms and other lab organisms has turned up many potential causes of aging. Some experts blame aging on the corrosive capability of chemically reactive oxygen molecules or “oxidants” churned out by mitochondria inside cells. DNA damage, including the shortening of chromosome endcaps (called telomeres) is also a prime suspect. Chronic, low-grade inflammation, which tends to get worse the older people get, wreaks so much havoc on tissues that some researchers believe it is aging’s prime cause, referring to aging as “inflammaging.” All these things and more have been proposed to be at the root of aging. Some researchers, like UCLA’s Steve Horvath, view aging as a biological program written on our DNA. He has seen evidence of a biological clock that marks milestones along life’s path. Some people reach those milestones more quickly than others, making them older biologically than the calendar suggests. Others take a more leisurely stroll, becoming biological youngsters compared with their chronological ages.
Many others, including Richard Miller, a geroscientist at the University of Michigan, deny that aging is programmed. Granted, a biological clock may measure the days of our lives, but it’s not a ticking time bomb set to go off on a particular date. After all, humans aren’t like salmon, which spawn, age and die on a schedule.
Instead, aging is a “by-product of running the engine of life,” says biodemographer Jay Olshansky of the University of Illinois at Chicago. Eventually bodies just wear out. That breakdown may be predictable, but it’s not premeditated. Despite all the disputes about what aging is or isn’t, scientists have reached one radical consensus: You can do something about it. Aging can be slowed (maybe even stopped or reversed). But exactly how to accomplish such a counterattack is itself hotly debated. Biotechnology and drug companies are developing several different potential remedies. Academic scientists are investigating many antiaging strategies in animal experiments. (Most of the research is still being done on mice and other organisms because human tests will take decades to complete.) Even researchers who think they have finally come up with real antiaging elixirs say they don’t have the recipe for immortality, though. Life span and health span, new research suggests, are two entirely separate things. Most researchers who work on aging aren’t bothered by that revelation. Their goal is not necessarily extending life span, but prolonging health span — the length of time people live without frailty and major diseases.
Aging as disease Many health problems are so commonly associated with aging that some researchers take the highly controversial stance that aging itself is a disease, says Saul Villeda of the University of California, San Francisco.
If aging is a disease, in Villeda’s lab it’s almost a contagious one: He can artificially spread aging from old lab mice to young ones. One mode of aging transmission is to give genetically identical mice transfusions of young or old blood. In another approach, researchers sew together pairs of mice so that their blood vessels will join up and link their circulatory systems.
This artificial joining of two separate animals, known as parabiosis, was a staple of physiology experiments for over a century before Irina Conboy got the idea to pair an old mouse with a young one. Conboy, a stem cell researcher at the University of California, Berkeley, made headlines with her experiments. Those headlines focused on the good news: Young blood rejuvenated old mice. In further studies by other researchers, infusions of young blood made broken bones in old mice heal better (SN Online: 5/19/15), gave their muscles extra spring and improved their memories (SN: 5/31/14, p. 8). Apparently some substances in the blood triggered the rejuvenation. Some candidates for those rejuvenation factors have been identified, although none are universally agreed on.
But news accounts mostly ignored the flip side of the experiment: Being tethered to an old mouse made young mice age faster. One substance in the blood of old mice, a protein called Beta‑2‑microglobulin, or B2M, seemed to prematurely age the young ones, Villeda and colleagues reported last year in Nature Medicine (SN: 8/8/15, p. 10). Parabiosis experiments don’t last very long, so no one knows whether youth or decrepitude will win in the end — or if the two mice would have settled into middle age together.
UCLA’s Horvath has evidence that the mice may never totally sync. He monitors aging by examining molecular tags called methyl groups, which attach to various locations on DNA in a process called methylation. Methylation is an epigenetic modification of DNA. Such modifications work something like flagging passages in a book with sticky notes. Attaching a tag doesn’t change the information in the book — it just draws attention to some passages and signals that others should be ignored.
Horvath measures DNA methylation changes at 353 different spots in the human genetic instruction book, or genome. As people age, 193 locations accumulate tags, like playbills plastered on urban buildings. At 160 others, methylation is gradually stripped away with age. Knowing how much methylation is normally found at each spot at a given chronological age allows Horvath to calculate biological age. Some people age at different rates than others, he discovered. For instance, semi-supercentenarians — people who live to be 105 to 109 — are about 8.6 years younger epigenetically than their chronological age. Their children are slow to age, too, though not as slow as their parents. Epigenetic clocks indicate that the offspring are about five years younger biologically than other people of the same chronological age.
People often joke about certain abilities, such as eyesight, memory or hearing being “the first to go.” Some of Horvath’s work suggests that the notion isn’t entirely far-fetched. He calculated the epigenetic age of specific organs and discovered that body parts can age at different rates. The cerebellum, the part of the brain that sits at the top of the brain stem and helps coordinate movement, speech and other activities, ages the slowest of the brain regions that Horvath analyzed. While there are natural differences in organ aging, some conditions, such as HIV infection and obesity, can prematurely age certain organs, Horvath and colleagues have found.
These experiments demonstrate that aging and its effects are malleable. “Aging is really plastic — it’s not set in stone,” says Conboy. Consequently, she and other researchers agree, something can be done to slow aging, or perhaps turn it around entirely. But exactly what can be done is vigorously disputed.
Interpret with caution Most scientists working on aging urge caution in extrapolating promising results in animal studies to humans. For instance, one of the most promising early candidates for a rejuvenation factor from young blood was a protein called GDF11. Reports in 2013 and 2014 concluded that GDF11 levels in blood decline with age; restoring the protein in old animals could reverse some heart problems, improve muscle strength and spur nerve cell growth in the brain. Since those reports, other researchers have disputed the protein’s revitalizing powers. In a recent study, researchers measured GDF11 levels in 140 people aged 21 to 93. Levels of the protein didn’t decline with age, Mayo Clinic researchers reported in the June 14 Cell Metabolism. Previous researchers may have gotten GDF11 mixed up with a similar protein called myostatin, which does dip as people get older. Not only does GDF11 not decline with age, having too much of it could be bad, the Mayo team found. People with higher blood levels of the protein were more likely to be frail, have diabetes and heart problems, and have a more difficult time recovering from surgery than people with lower levels of the protein.
Beyond the blood experiments, scientists have examined various ideas about what goes wrong in aging and have devised strategies to counteract it. For instance, some evidence suggests that stem cells run out of steam as they get older. Restoring old stem cells to youthful vigor may enable them to repair or replace damaged tissues and turn back the biological clock. Keeping stem cells youthful may involve sheltering them from inflammation or things that could damage their DNA.
One way to keep stem cells and other cells working is to avoid the loss of telomeres capping the ends of chromosomes. As cells divide, their telomeres grow shorter until they are so short that chromosomes can no longer safely replicate. That may be a signal for the cell to shut down or die. So some researchers think that lengthening telomeres could give cells the protection needed to survive longer.
One biotechnology company executive flew from the United States to Colombia to try out her company’s gene therapy for lengthening telomeres. That decision bypassed U.S. government and other safety measures designed to protect human study participants. And no one knows whether it will work or doom her to cancer, which often relies on long telomeres to keep growing.
Other researchers are exploring more measured approaches to antiaging therapies. One study in dogs is testing rapamycin, the first drug shown to lengthen mouse life spans. Rapamycin is an immune suppressant that also has anticancer effects. The rationale for using it came from research on caloric restriction, the world-champion method for making animals live longer. Animals on calorie-restricted diets typically eat at least 25 percent fewer calories than normal. Such low-cal treatment has increased life spans in mice, dogs, fruit flies, yeast, worms and other lab organisms. Results from primate studies have been mixed (SN: 8/1/09, p. 9; SN: 10/6/12, p. 8). Some people have put themselves on caloric-restriction regimes (SN: 10/25/08, p. 17). A handful of studies suggest that those people have better health, but it’s too soon to know whether they will outlive their peers.
Exactly why drastically reducing food intake can extend life isn’t known. But researchers have good evidence that a series of biochemical reactions known as the mTOR pathway is involved. The protein mTOR helps monitor nutrient levels in cells and regulates cell movement, protein production, and cell growth and survival. When starvation sets in, cells turn off mTOR’s activity, which allows a self-cannibalizing process called autophagy to scavenge nutrients by digesting some of the cell’s internal organs. This internal garbage disposal and recycling method also removes old, worn-out mitochondria and proteins that may otherwise keep cells from functioning efficiently. That process and other cellular activities governed by mTOR may be responsible for making cells, and organisms, live longer. Rapamycin gave mTOR its name — mechanistic target of rapamycin. Giving the drug might do what caloric restriction does without requiring superstrict diets (SN: 6/4/11, p. 22). Matt Kaeberlein, a geroscientist at the University of Washington, and colleagues conducted a safety study of the drug last year in 24 dogs. The study was only 10 weeks long, so the researchers can’t yet draw any conclusions about long-term effects on aging. But the dogs had no major side effects from taking low doses of the drug, a worry because rapamycin impairs immune system function and could make animals (including people) who take it more vulnerable to infection or cancer.
Rapamycin’s drawbacks make it unattractive for human studies. The diabetes drug metformin may instead be the antiaging drug of choice for people, says gerontologist Nir Barzilai. In addition to mTOR, metformin targets an insulin-like growth protein known as IGF-1. That protein has been implicated in a variety of biological processes that promote aging.
Barzilai, of Albert Einstein College of Medicine in New York City, and researchers at more than a dozen centers around the country plan to test metformin for its ability to fight aging in people 65 to 79. Barzilai and colleagues laid out the case for using metformin in the June 14 Cell Metabolism. Metformin is generally safe, with few major side effects. It has been shown to improve a variety of health measures and to impair cancer development in people with type 2 diabetes (SN: 11/30/13, p. 18). Barzilai says the drug may help people who don’t have diabetes also live healthier when they are elderly. If it does, commercials touting metformin might have to add another disclaimer, he jokes. “The commercials will go on: ‘This will make you healthy, but we have to apologize because you might live longer.’ ”
But studies of mice suggest that disclaimer may not be necessary. Research by Miller and others suggests that metformin may not prolong life. They have been dosing mice with various chemicals, including metformin and rapamycin, looking for drugs that will make mice healthier and live longer. In a new study, published online June 16 in Aging Cell, Miller and colleagues fed mice metformin starting when the rodents were 9 months old — middle age for a mouse. Combining metformin and rapamycin didn’t make the mice live much longer than rapamycin alone did in previous trials.
Cellular zombies Other researchers are hoping to stave off death by getting rid of the undead. Cellular zombies called senescent cells are stressed cells that have entered a type of stasis — they’re not dead, but they’re not functioning either. Stress for cells usually means severe DNA damage that could produce cancer, critically short telomeres or other molecular catastrophes that trigger shutdown mode. That lockdown is for the greater good, says aging researcher Judith Campisi, who studies senescence at the Buck Institute for Research on Aging in Novato, Calif. “It’s protective,” she says. “You don’t want defective cells to propagate.” (When damaged cells continue to grow they may become cancerous.)
Unfortunately, says Campisi, the senescent cells don’t die. Instead they send out messages to neighboring cells: “Hey, there’s a problem. Be prepared. What happened to me could happen to you.” Such messages are probably intended as public service announcements, but they could trigger mass panic and inflammation. Like zombies putting the bite on the living, senescent cells damage surrounding cells and accelerate aging.
Researchers have worked out methods for hitting the zombie cells with genetic shots to the head, effectively destroying the cells and removing them from the body. Mice from which senescent cells have been removed had increased median life span and improved health, researchers reported in Nature in February (SN: 3/5/16, p. 8).
Campisi and other researchers are working on ways to clear senescent cells from humans, too. But no antiaging treatment makes mice or any other animal live forever. Researchers have yet to increase a mouse’s life span (which rarely goes above two years) to five years, although one mouse fell just short of that mark.
Much research suggests that things that extend life span, such as rapamycin, might not stretch health spans. Mutations that make millimeter-long transparent worms known as Caenorhabditis elegans live longer also extend the proportion of their lives the worms spend being frail, Heidi Tissenbaum of the University of Massachusetts Medical School in Worcester and colleagues reported last year in the Proceedings of the National Academy of Sciences.
But living healthy doesn’t guarantee longevity either, a new study of sea urchins suggests. Red sea urchins (Mesocentrotus franciscanus) live well past 100 years old in the wild, while purple sea urchins (Strongylocentrotus purpuratus) make it to 50. But variegated (also called “green”) sea urchins (Lytechinus variegatus) normally die after four years. The difference in the species’ life spans might be due to different rates of aging, thought aging researcher Andrea Bodnar at the Bermuda Institute of Ocean Sciences in St. George’s and developmental biologist James Coffman of the MDI Biological Laboratory in Salisbury Cove, Maine. Instead, they found, none of the species seem to age at all . Young and old members of each species are similar in their abilities to reproduce and to regenerate spines and tube feet, the researchers reported online April 20 in Aging Cell . Even though the short-lived variegated urchins have no signs of slowing down, they still die. Why is a mystery, Coffman says. Ways to be wellderly A similar paradox is also seen in “wellderly” people that geneticist Ali Torkamani has been studying at the Scripps Research Institute in La Jolla, Calif. About eight years ago, Torkamani started bringing in people over 80 who had made it to an advanced age without any sign of chronic disease. The idea was to study their DNA and learn the secrets of healthy aging.
Despite living healthy, the wellderly didn’t carry genetic variants connected with extremely long lives, Torkamani and colleagues discovered. The wellderly also had no genetic advantage when it comes to cancer, stroke or diabetes. What they did have was a lower risk of getting Alzheimer’s and heart disease. Each of the wellderly seemed to have their own genetic recipe for success, suggesting there are lots of ways to stay healthy into old age. The researchers didn’t rule out that diet and lifestyle also help. “There’s hope for everybody,” Torkamani declares.
But his cloud of optimism may have a tarnished lining. His findings, along with the sea urchin and worm results, suggest that aging and longevity aren’t the same things. If that’s the case, it would mean that stopping aging would not extend human life span by much. The oldest (verified) person to have ever lived was Jeanne Louise Calment, a French woman who died at age 122 in 1997. People might top out at 130 if aging is controlled (and most people still would not make it that long because they just don’t have the necessary makeup). As a species, humans probably can’t go further without changing whatever controls longevity too, some researchers think.
Exactly how long people can live won’t be answered until proven antiaging therapies are developed. If aging and longevity are linked, then treating aging could very well make people live longer, healthier lives. If they are separate phenomena, then people could forgo the cancer, heart disease and other ailments of aging, but they would still have limited life spans. In that case Star Trek’s Mr. Spock might need to revise his usual parting words. When talking to humans, he should wish that they will live long or prosper. We may not get both.
Happy 40th anniversary, Viking 1! Four decades ago — July 20, 1976 — the robotic probe became the first U.S. mission to land on Mars. Its sister spacecraft, Viking 2, touched down 45 days later.
Launched August 20, 1975, Viking 1 spent over 6 years snapping pictures and studying the soil at its landing site, an ancient crater named Chryse Planitia. An experiment to look for Martian microbes turned up nothing definitive, though some researchers continue to argue otherwise.
Viking 1 wasn’t the first to successfully touch down on the Red Planet. That honor goes to the Soviet probe Mars 3, whichgently landed on Mars in 1971, though its only transmission — a partial, garbled image — lasted just 20 seconds.
Today, seven probes actively call Mars home. A European-led orbiter and lander, ExoMars, is on its way, and NASA has two missions lined up: the Insight lander, whose launch was recently delayed to 2018, and the Mars 2020 rover, which will pick up where the Vikings left off and search for Martian life.
Microbes may have played a role in making us, us. A new study shows similar patterns in the evolution of gut bacteria and the primates they live in, suggesting that germs and apes could have helped shaped one another.
For at least 10 million years, bacteria have been handed down from the common ancestor of humans and African apes. As apes split into separate species, so did the microbes inside them, researchers report July 22 in Science. Now, relationships between gut bacterial species mirror the family tree of gorillas, humans, bonobos and chimpanzees. Germs are a piece of our history, says evolutionary biologist Andrew Moeller who led the study while at both the University of Texas at Austin and the University of California, Berkeley. “Just like genes we’ve inherited from our ancestors,” he says, “we’ve inherited some of our bacteria from our ancestors as well.”
It’s well known that bacteria are key to human health (SN: 04/02/16, p. 23). They play major roles in the immune system and development. But very few researchers have turned to the past, Moeller says, to ask how humans got those handy bacteria in the first place. His team studied three families of bacteria living in the feces of people from Connecticut, as well as in that of wild chimps, bonobos and gorillas. The scientists used DNA evidence to build relationship trees for each bacterial family, then compared each tree with known relationships between humans and close primate relatives.
Two of three bacterial trees matched primate relationships. For those families, closely related bacteria live in closely related primates. For humans, “the closest relatives of our gut bacteria live in chimpanzees,” Moeller says, “just like our closest relatives are chimps.”
Scientists would expect that pattern to match only if apes and bacteria split into new species in unison. The fact that apes and bacteria split at roughly the same time, while bacteria were living inside of ape species, implies that they were influencing each other, and therefore that the evolution of one group could shape the evolution of the other.
Changing bacteria may have “allowed us to evolve,” says microbial geneticist Julia Segre of the National Human Genome Research Institute in Bethesda, Md., who was not involved in the new work. She and conservationist Nick Salafsky of the nonprofit Foundations of Success, also in Bethesda, wrote a perspective on it in the same issue of Science.
A “very intimate relationship with bacteria,” she says, “is part of who we are.” While the researchers agree that humans and bacteria probably shaped each other’s evolution, they caution that it’s too soon to tell if (and how) ancient apes and microbes changed each other.
Those ancient relationships may get harder to study over time. Industrialization and antibiotics have reduced the diversity of bacteria living in and on humans, Moeller says. And while the microbes in this study have stuck around, other groups may have disappeared or changed dramatically.
One caveat, Segre says, is that humans have been exposed to antibiotics and modern life. Wild African apes might still have their ancient gut flora, but people in Connecticut might not (SN: 12/13/14, p. 10). It’s especially important to do studies like this now, she says, “because it’s not going to get better.”
In the future, Moeller says, researchers should look deeper into the past to see if the gut bacteria living in all mammals share one common ancestor. Scientists could also go the other way, he says, to see if more recently divided human populations also have characteristic gut bacteria.
How can aging be delayed? How does the brain age? And what does aging look like in animals, plants or the rest of the natural world? The July 23 issue of Science News tackles these questions and more in a special report called “Aging’s Future.”
On Tuesday, July 26, at 3 p.m. EDT, three Science News reporters will answer questions about aging as part of Reddit’s Ask Me Anything series. Molecular biology reporter Tina Hesman Saey, neuroscience writer Laura Sanders and biology writer Susan Milius will be responding to questions from 3 p.m. to 4 p.m. Eastern at this link.
Read their in-depth features on aging:
A healthy old age may trump immortality: Despite disagreements about what aging is and isn’t, scientists have reached a radical consensus: It can be delayed. By Tina Hesman Saey
The brain’s blueprint for aging is set early in life: The brain’s decline may mirror its beginning, offering clues to aging. By Laura Sanders
Organisms age in myriad ways — and some might not even bother: There is great variety in how animals and plants deteriorate (or don’t) over time. By Susan Milius